


理化学研究所
静岡県立総合病院
欧洲杯押注平台_2024欧洲杯网站-官网app
理化学研究所(理研)生命医科学研究センターゲノム解析応用研究チームの小杉俊一研究員(研究当時、現客員研究員、静岡県立総合病院リサーチサポートセンター遺伝研究部研究員)、薬学部ゲノム病態解析講座の寺尾知可史特任教授(理化学研究所生命医科学研究センター ゲノム解析応用研究チーム チームリーダー、静岡県立総合病院 臨床研究部免疫研究部長)らの共同研究グループは、全ゲノムシークエンス[1]データから「構造多型(SV)[2]」を高精度で検出する新しい手法を開発しました。
本研究成果は、これまで発見できなかった疾患や形質の原因となる遺伝子やゲノム変異の同定に貢献すると期待できます。
SVとは、個人間のゲノムの違いのうち50塩基対以上の長さの変異のことです。これまでSVを検出する多くのツールが開発されてきましたが、精度よく検出できる単独のツールは存在しませんでした。
今回、共同研究グループは既存の複数のツールを用いてSVを高精度に選別し、この選別過程で抜け落ちたSVを独自の遺伝子型[3]判定手法により回収する、新しいSV検出手法「MOPline」を開発しました。MOPlineを用いて、バイオバンク?ジャパン(BBJ)[4]に登録された約3,300人の全ゲノムシークエンスデータから約134,000(個人当たり約16,000)のSVを検出し、このSVと約18万人のBBJデータを用いて解析をしたところ、多くの疾患や量的形質[5]にSVが関わっていることが明らかになりました。
本研究は、科学雑誌『Cell Genomics』オンライン版(5月18日付:日本時間5月19日)に掲載されました。
静岡県立総合病院
欧洲杯押注平台_2024欧洲杯网站-官网app
理化学研究所(理研)生命医科学研究センターゲノム解析応用研究チームの小杉俊一研究員(研究当時、現客員研究員、静岡県立総合病院リサーチサポートセンター遺伝研究部研究員)、薬学部ゲノム病態解析講座の寺尾知可史特任教授(理化学研究所生命医科学研究センター ゲノム解析応用研究チーム チームリーダー、静岡県立総合病院 臨床研究部免疫研究部長)らの共同研究グループは、全ゲノムシークエンス[1]データから「構造多型(SV)[2]」を高精度で検出する新しい手法を開発しました。
本研究成果は、これまで発見できなかった疾患や形質の原因となる遺伝子やゲノム変異の同定に貢献すると期待できます。
SVとは、個人間のゲノムの違いのうち50塩基対以上の長さの変異のことです。これまでSVを検出する多くのツールが開発されてきましたが、精度よく検出できる単独のツールは存在しませんでした。
今回、共同研究グループは既存の複数のツールを用いてSVを高精度に選別し、この選別過程で抜け落ちたSVを独自の遺伝子型[3]判定手法により回収する、新しいSV検出手法「MOPline」を開発しました。MOPlineを用いて、バイオバンク?ジャパン(BBJ)[4]に登録された約3,300人の全ゲノムシークエンスデータから約134,000(個人当たり約16,000)のSVを検出し、このSVと約18万人のBBJデータを用いて解析をしたところ、多くの疾患や量的形質[5]にSVが関わっていることが明らかになりました。
本研究は、科学雑誌『Cell Genomics』オンライン版(5月18日付:日本時間5月19日)に掲載されました。
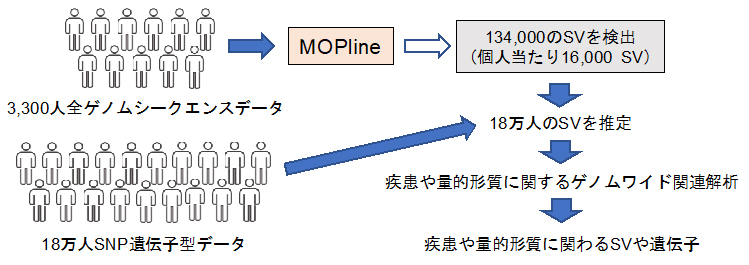
構造多型(SV)を用いて疾患や量的形質に関わるSVや遺伝子を究明
背景
ゲノムの「構造多型(SV)」は、50塩基対(bp)以上の欠失[6]、挿入[7]、重複[8]、逆位[9]多型の総称であり、50bpより小さい欠失、挿入に相当する「インデル」および1bpの塩基置換である「一塩基多型(SNV)」[10]とは区別されます。SVの出現頻度は個人当たり1万~2万と、インデル(個人当たり約70万)やSNV(個人当たり約400万)に比べて低いものの、サイズが大きいために、SVに起因する個人ゲノム間の異なる塩基数は、SNVによる違いの塩基数の3~10倍あることが示されています。
このように個人ゲノム間に大きな違いをもたらすSVは、発達障害や知的障害を含むさまざまなヒトの疾患?形質の遺伝的要因となることが近年の多くの研究から示されています注1、2)。また、がんなどの体細胞変異によって引き起こされる疾患においても、SVが関わることを示す多くの研究があります注3、4)。
一方で、SVの構造の複雑さと大きいサイズのために、SVの検出はSNVと比較して困難です。ゲノムの多型は通常、100~150bpの短い配列(リード[11])データをヒトの標準ゲノム配列(リファレンス配列[12])にアライメント[13]して検出します。このリード長内に収まるSNVやインデルに対して、より大きなサイズのSVはリード内に収まらず、SVをまたいでアライメントされるリードの間接的な証拠を用いて検出しなければならないため、検出精度(検出の正確性)や検出感度(検出の効率)が低くなってしまいます。これまでに多くのSV検出ツールが開発されてきましたが、検出結果の共通性が低いという問題があり、単一のツールで精度、感度ともに高くSVを検出できるツールは存在しませんでした注5)。
注1)Weischenfeldt J, et al. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125-38 (2013).
注2)Marshall, C.R. et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27-35 (2017).
注3)Yi, K. et al. Patterns and mechanisms of structural variations in human cancer. Exp. Mol. Med. 50, 98 (2018).
注4)Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47-54 (2016).
注5)Kosugi, S. et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 20, 117 (2019).
このように個人ゲノム間に大きな違いをもたらすSVは、発達障害や知的障害を含むさまざまなヒトの疾患?形質の遺伝的要因となることが近年の多くの研究から示されています注1、2)。また、がんなどの体細胞変異によって引き起こされる疾患においても、SVが関わることを示す多くの研究があります注3、4)。
一方で、SVの構造の複雑さと大きいサイズのために、SVの検出はSNVと比較して困難です。ゲノムの多型は通常、100~150bpの短い配列(リード[11])データをヒトの標準ゲノム配列(リファレンス配列[12])にアライメント[13]して検出します。このリード長内に収まるSNVやインデルに対して、より大きなサイズのSVはリード内に収まらず、SVをまたいでアライメントされるリードの間接的な証拠を用いて検出しなければならないため、検出精度(検出の正確性)や検出感度(検出の効率)が低くなってしまいます。これまでに多くのSV検出ツールが開発されてきましたが、検出結果の共通性が低いという問題があり、単一のツールで精度、感度ともに高くSVを検出できるツールは存在しませんでした注5)。
注1)Weischenfeldt J, et al. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125-38 (2013).
注2)Marshall, C.R. et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27-35 (2017).
注3)Yi, K. et al. Patterns and mechanisms of structural variations in human cancer. Exp. Mol. Med. 50, 98 (2018).
注4)Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47-54 (2016).
注5)Kosugi, S. et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 20, 117 (2019).
研究手法と成果
全ゲノムシークエンスデータから高い信頼度(高精度)を持つSVを取得する方法の一つは、既存のSV検出ツール間で共通に検出されるSV(ツール間オーバーラップSV)を選別することです。しかし共同研究グループは、必ずしもツール間オーバーラップSVが高い精度を示すわけではないことを見いだしました。
そこで、オーバーラップSVが高い精度を示す既存ツールの組み合わせを調べました。そして、既存の4~9個のツールを用いて最適なツールの組み合わせをSVタイプやサイズごとに決定するアルゴリズムを開発し、MOP(Merging Overlap calls from selected Pairs of algorithms)と名付けました(図1上)。
MOPを用いると、高い精度を持ったSVを選別できますが、一部のSVは見逃してしまいます。この問題を解決するために、MOPでSVが検出されなかったゲノム領域をスキャンし、SVの存在を確認する作業を行いました。この存在確認では、リードのアライメント情報を用いた独自の遺伝子型判別手法を用い、このSVの再判別手法をSMC(Supplementing Missing Calls)と名付けました(図1下)。そして最終的に、MOP、SMC、およびフィルタリングやアノテーション[14]機能を組み合わせたSV検出手法、「MOPline」の開発に成功しました(図1)。
そこで、オーバーラップSVが高い精度を示す既存ツールの組み合わせを調べました。そして、既存の4~9個のツールを用いて最適なツールの組み合わせをSVタイプやサイズごとに決定するアルゴリズムを開発し、MOP(Merging Overlap calls from selected Pairs of algorithms)と名付けました(図1上)。
MOPを用いると、高い精度を持ったSVを選別できますが、一部のSVは見逃してしまいます。この問題を解決するために、MOPでSVが検出されなかったゲノム領域をスキャンし、SVの存在を確認する作業を行いました。この存在確認では、リードのアライメント情報を用いた独自の遺伝子型判別手法を用い、このSVの再判別手法をSMC(Supplementing Missing Calls)と名付けました(図1下)。そして最終的に、MOP、SMC、およびフィルタリングやアノテーション[14]機能を組み合わせたSV検出手法、「MOPline」の開発に成功しました(図1)。
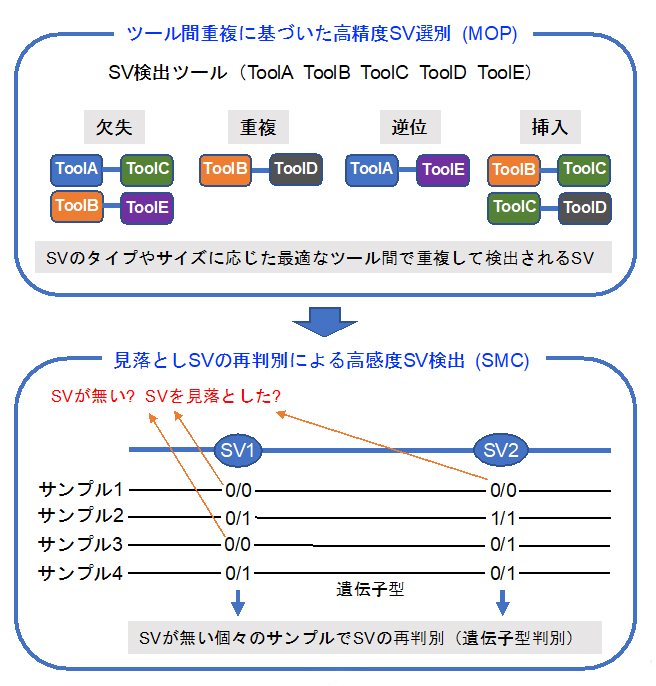
図1 今回開発した「MOPline」のアルゴリズム
構造多型(SV)のタイプやサイズに応じて最適なツールの組み合わせを選別し、精度の高いツール間共有SVを選別(MOP)。次にSMCアルゴリズムを用いて見落としたSVを検出し、検出感度を向上させる。
MOPlineのSV検出精度、検出感度をNA12878などの全ゲノムシークエンスデータを用いて検証したところ、MOPlineは既存のツールの精度、感度を上回っていました。さらに、複数のツールを組み合わせてSVを検出する既存のパイプライン(GATK-SV、sv-pipeline)との比較を公共データベース(1000人ゲノムプロジェクト[15])から取得した100の全ゲノムシークエンスデータを用いて行いました。その結果、MOPlineのSV検出精度はGATK-SVのものと同等ながら、真陽性SV(特に挿入)の検出数(検出感度)に関してはGATK-SVおよびsv-pipelineを上回っていました(図2)。
MOPlineのSV検出精度、検出感度をNA12878などの全ゲノムシークエンスデータを用いて検証したところ、MOPlineは既存のツールの精度、感度を上回っていました。さらに、複数のツールを組み合わせてSVを検出する既存のパイプライン(GATK-SV、sv-pipeline)との比較を公共データベース(1000人ゲノムプロジェクト[15])から取得した100の全ゲノムシークエンスデータを用いて行いました。その結果、MOPlineのSV検出精度はGATK-SVのものと同等ながら、真陽性SV(特に挿入)の検出数(検出感度)に関してはGATK-SVおよびsv-pipelineを上回っていました(図2)。
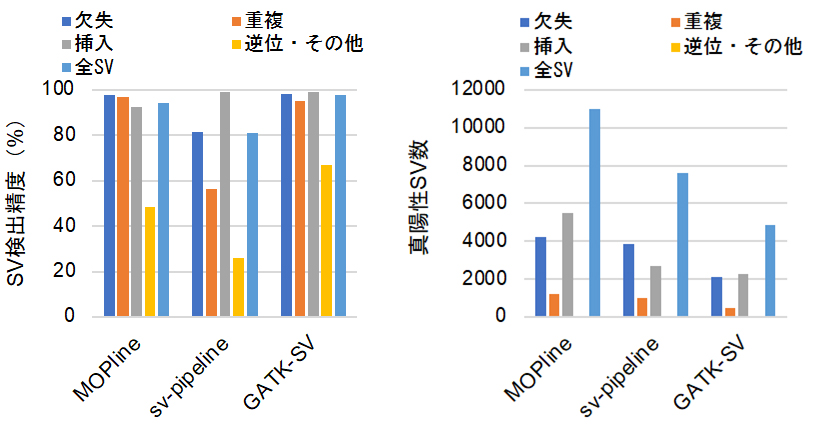
図2 MOPlineと複数ツールを用いる既存パイプラインとの性能比較
100サンプルのSVを対象として、それぞれの方法の検出精度をロングリードデータを用いて決定した。MOPlineのSV検出精度は逆位以外で約94%以上を示し、GATK-SVとほぼ同等だった(左)。一方、真陽性検出数については、MOPlineが三つの中で最も高かった(右)。
次に、MOPlineを用いて3,258人のバイオバンク?ジャパン(BBJ)全ゲノムシークエンスデータからSVを検出しました。その結果、約134,000(個人当たり約16,000)のSVが検出され、この数はこれまでの大規模SV研究プロジェクトで検出された個人当たりのSV数の1.7~3.3倍高いものでした(図3)。
次に、MOPlineを用いて3,258人のバイオバンク?ジャパン(BBJ)全ゲノムシークエンスデータからSVを検出しました。その結果、約134,000(個人当たり約16,000)のSVが検出され、この数はこれまでの大規模SV研究プロジェクトで検出された個人当たりのSV数の1.7~3.3倍高いものでした(図3)。
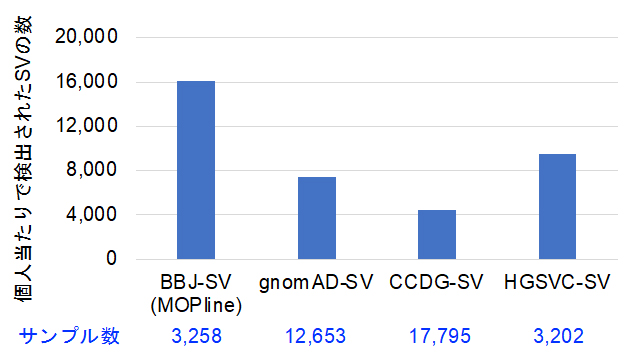
図3 MOPlineを用いてBBJ全ゲノムシークエンスデータから検出された個人当たりのSV数
MOPlineを用いた場合と他の三つの大規模SV研究プロジェクトであるgomoAD-SV (Collins et al., Nature 2020)、CCDG-SV (Abel et al., Nature 2020)、HGSVC-SV (Byrska-Bishop et al., Cell 2022)で検出された個人あたりのSV数の比較。MOPlineを用いた場合は、他の三つのプロジェクトの1.7~3.3倍に達した。
このBBJ全ゲノムシークエンスデータは、がんや認知症などのうち少なくとも一つの疾患を持つ患者からのものでした。そこで、疾患に関わる既知遺伝子のタンパク質コーディング領域と重なるSVを調べたところ、いくつかのまれに存在するSVが、疾患サンプルに特異的な既知の疾患リスク遺伝子(大腸がん、乳がんなど)のタンパク質コード領域と重なっていることが分かりました(表1)。
このBBJ全ゲノムシークエンスデータは、がんや認知症などのうち少なくとも一つの疾患を持つ患者からのものでした。そこで、疾患に関わる既知遺伝子のタンパク質コーディング領域と重なるSVを調べたところ、いくつかのまれに存在するSVが、疾患サンプルに特異的な既知の疾患リスク遺伝子(大腸がん、乳がんなど)のタンパク質コード領域と重なっていることが分かりました(表1)。
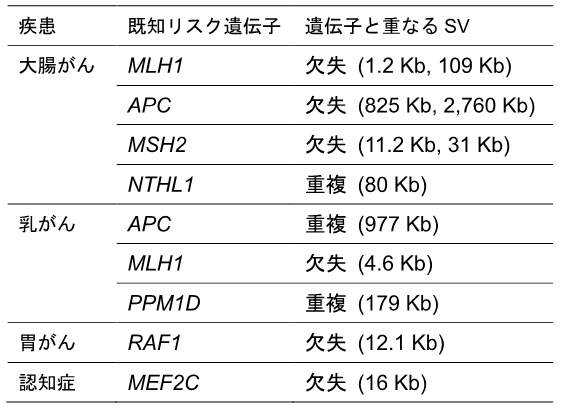
表1 BBJ-SVデータに見いだされた既知疾患リスク遺伝子コード領域と重なるまれなSVの例
4種類の疾患に関わるまれな12個のSVが、既知疾患リスク遺伝子のタンパク質コード領域と重なっていた。
MOPlineで検出されたBBJ-SV(約134,000)を参照パネル[16]として用い、18万人のSNPアレイデータ[17](SNP遺伝子型データ)のインピュテーション[16]を行い、約18万人のSVを類推しました。類推したSVと約18万人の医療情報を用い、42の疾患と60の量的形質に対するゲノムワイド関連解析(GWAS)[18]を行いました。その結果、がんなどの疾患を含む32形質に関して、SNPと同等もしくはより強い相関を示す41のSVが見いだされました。相関のあったSVのうち、8個のSVは関連遺伝子のコード領域と重なっており、そのうちの5個(MUC22、APOC1、GYPA/GYPB、RP11-219A15、FUT2に重なるSV)は、これまでに該当形質との関連の報告が無い新しく同定されたSVでした(表2)。
MOPlineで検出されたBBJ-SV(約134,000)を参照パネル[16]として用い、18万人のSNPアレイデータ[17](SNP遺伝子型データ)のインピュテーション[16]を行い、約18万人のSVを類推しました。類推したSVと約18万人の医療情報を用い、42の疾患と60の量的形質に対するゲノムワイド関連解析(GWAS)[18]を行いました。その結果、がんなどの疾患を含む32形質に関して、SNPと同等もしくはより強い相関を示す41のSVが見いだされました。相関のあったSVのうち、8個のSVは関連遺伝子のコード領域と重なっており、そのうちの5個(MUC22、APOC1、GYPA/GYPB、RP11-219A15、FUT2に重なるSV)は、これまでに該当形質との関連の報告が無い新しく同定されたSVでした(表2)。
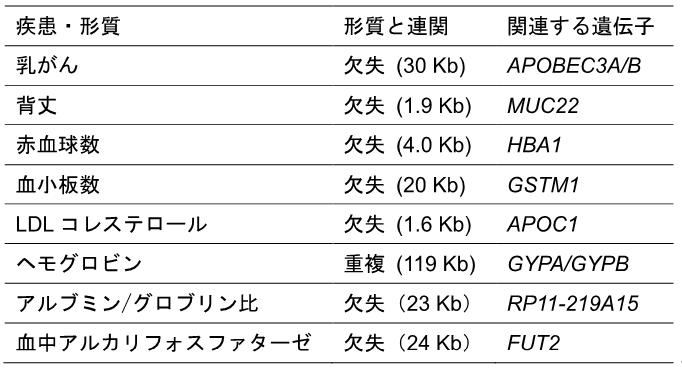
表2 ゲノムワイド関連解析(GWAS)で見いだされた疾患?量的形質と相関するSVの例
がんなどの疾患を含む32形質について、SNPと同等もしくはそれ以上の強い相関を示す41のSVのうち、関連遺伝子のコード領域と重なる8個を掲載した。乳がんを除く量的形質に関わるSVのうち、ヘモグロビンの重複および血中アルカリフォスファターゼに関わる欠失はマイナスの方向(減少する)に働き、他の形質に関わるSVはプラスの方向(増加する)に作用する。
以上の研究結果は、MOPlineはこれまでにないSV検出精度と検出感度を示すツールであり、単一遺伝子疾患の原因となるまれなSVの同定を可能にするだけでなく、SVのインピュテーションを行うことで複雑な量的形質に関わるSVの同定を可能にすることを示しています。
以上の研究結果は、MOPlineはこれまでにないSV検出精度と検出感度を示すツールであり、単一遺伝子疾患の原因となるまれなSVの同定を可能にするだけでなく、SVのインピュテーションを行うことで複雑な量的形質に関わるSVの同定を可能にすることを示しています。
今後の期待
本研究成果は、これまで主にSNPを用いて行われていた疾患に関わるゲノム解析を、構造多型を含めた解析に拡張させることを可能にします。
また、MOPlineは、単一および数千の全ゲノムシークエンスデータを用いたSV検出を可能とし、ヒトを含む多様な生物種のSVを検出することができます。このことから、幅広い研究分野において、これまで既存のツールでは不可能であったSVの研究が可能となると期待できます。
また、本研究で得られた日本人3,253人のBBJ-SVデータは、これまでにない高精度な大規模データであり、SVの検証やインピュテーションのための貴重な研究資源として活用されることが期待できます。
また、MOPlineは、単一および数千の全ゲノムシークエンスデータを用いたSV検出を可能とし、ヒトを含む多様な生物種のSVを検出することができます。このことから、幅広い研究分野において、これまで既存のツールでは不可能であったSVの研究が可能となると期待できます。
また、本研究で得られた日本人3,253人のBBJ-SVデータは、これまでにない高精度な大規模データであり、SVの検証やインピュテーションのための貴重な研究資源として活用されることが期待できます。
論文情報
<タイトル>
Detection of trait-associated structural variations using short read sequencing
<著者名>
Shunichi Kosugi, Yoichiro Kamatani, Katsutoshi Harada, Kohei Tomizuka, Yukihide Momozawa, Takayuki Morisaki, The Biobank Japan Project, and Chikashi Terao
<雑誌>
Cell Genomics
<DOI>
10.1016/j.xgen.2023.100328
(クリックすると出版社サイトへ移動します)
Detection of trait-associated structural variations using short read sequencing
<著者名>
Shunichi Kosugi, Yoichiro Kamatani, Katsutoshi Harada, Kohei Tomizuka, Yukihide Momozawa, Takayuki Morisaki, The Biobank Japan Project, and Chikashi Terao
<雑誌>
Cell Genomics
<DOI>
10.1016/j.xgen.2023.100328
(クリックすると出版社サイトへ移動します)
補足説明
[1] 全ゲノムシークエンス
次世代シークエンス技術または第三世代シークエンス技術を用いて、全ゲノムDNAを鋳型として配列を解読すること。この配列解読によって、全ゲノム長の数倍~数十倍の総塩基数に相当するショートリードまたはロングリードデータが生成される。構造多型の検出には、ショートリードでゲノム長の10~30倍、ロングリードで10倍以上の全ゲノムシークエンスデータを要する。
[2] 構造多型(SV)
ゲノムの個人間の違いのうち、50bp以上の大きさの変異。変異のパターンに応じて、欠失、挿入、重複、逆位、転座などに分類されるが、これらが混在した複雑なパターンを示す構造多型も存在する。通常、塩基対数の小さい構造多型ほど数が多いが、染色体レベルで起こる大きなサイズの構造多型も存在する。SVはStructural Variationの略。
[3] 遺伝子型
ある遺伝子座で個人が持つ遺伝子変異のこと。どちらかの親から一つの変異を受け継いでいる場合ではヘテロ遺伝子型となり、両親から同じ変異を受け継いでいる場合にはホモ遺伝子型となる。
[4] バイオバンク?ジャパン(BBJ)
日本人集団27万人を対象とした生体試料のバイオバンクで、東京大学医科学研究所内に設置されている。理化学研究所が取得した約20万人のゲノムデータを保有する。オーダーメイド医療の実現プログラムを通じて実施され、ゲノムDNAや血清サンプルを臨床情報とともに収集し、研究者へのデータ提供や分譲を行っている。
[5] 量的形質
身長や体重など連続的、量的に変化する形質。疾患の有無などのbinary形質と区別される。
[6] 欠失
構造多型のタイプの一つで、ゲノム配列の一部が失われた形態。挿入と並び、構造多型の中では最も多く存在する。
[7] 挿入
構造多型のタイプの一つで、ゲノム配列の特定の位置に別の配列が挿入された形態。挿入配列で最も多くあるものが、内在レトロ因子が挿入されたタイプで、ミトコンドリア配列やウイルスゲノム配列が挿入されたタイプもある。欠失と並び、構造多型の中では最も多く存在する。
[8] 重複
構造多型のタイプの一つで、ゲノム配列の一部の領域が重複して(2コピー以上)挿入された形態。欠失や挿入に比べて数は少ないが、重複された領域内に遺伝子が含まれる場合、通常と異なる遺伝子発現パターンを示すことが多いため、遺伝子機能の喪失を引き起こす欠失と同様、疾患との関わりが多く報告されている。
[9] 逆位
構造多型のタイプの一つで、ゲノム配列の一部が通常と逆方向に変換されている形態。構造多型のタイプの中で最も数が少ない。
[10] 一塩基多型(SNV)
ゲノムの個人間の違いのうち、塩基配列上の1カ所の違い(置換)が一塩基多型と定義される。SNVはSingle nucleotide variantの略。SNVのうち、ある集団内での頻度が1%以上あるものをSNP(Single Nucleotide Polymorphism)と呼ぶ。
[11] リード
DNAの配列決定(シークエンシング)によって得られるDNA断片の配列情報。次世代シークエンシング技術で得られるリードは、通常100~200bpのショートリード断片であり、ヒトゲノム解読の場合、数億~10億本のリードを得る。第三世代シークエンシング技術では、平均7~10Kb(7,000~10,000bp)のロングリードが得られる。
[12] リファレンス配列
ある生物種のゲノム配列として、標準ゲノム配列として公開されているもの。ヒトでは、hg19やGRCh37などの総塩基数約3Gb(30億b)のゲノムリファレンスが公開されている。リファレンスにリードデータをアライメントすることにより、標準リファレンス配列と異なるDNA多型が検出される。
[13] アライメント
シークエンスリードをリファレンス配列上の合致する位置に対応付けすること。通常、ショートリードはbwaなどのアライメントツールを用いてアライメントし、得られたアライメントファイルを用いて構造多型を検出する。
[14] アノテーション
ゲノム上の遺伝子領域などに注釈付けを行うこと。遺伝子がどのゲノム位置にコードされているか、SVなどのゲノム変異がどの遺伝子領域と重なっているかなどの注釈付けがある。
[15] 1000人ゲノムプロジェクト
ヒトゲノムの遺伝的多様性を明らかにすることを目標として開始された国際共同研究プロジェクト。現在では2,500人以上のゲノム解析へ移行している。
[16] 参照パネル、インピュテーション
ヒトの場合、DNAマイクロアレイを用いて得られたデータには数十万のSNP情報が含まれるが、実際には数百万以上のSNPが存在する。アレイに含まれないSNPやインデルを推定する手法がインピュテーションである。通常多くのサンプルの全ゲノムシークエンスデータから得られたSNP遺伝子型情報を参照パネルとして、そのSNP遺伝子型の並び情報を基に遺伝子型推定を行う。
[17] SNPアレイデータ
SNP(Single Nucleotide Polymorphism)は一塩基多型(SNV)のうち、ある集団内での頻度が1%以上あるものを指す。SNPアレイデータは、DNAマイクロアレイを用いて得られるSNP遺伝子型データのこと。全ゲノムシークエンスデータを用いて得られるSNP遺伝子型よりも少ない遺伝子型情報しか得られないが、全ゲノムシークエンスよりも安価に解析できる。
[18] ゲノムワイド関連解析(GWAS)
疾患などの特定の形質を持った集団と持たない集団との間でSNP遺伝子型の頻度差の有意性を統計的に評価する手法。ゲノム全域にわたって一つ一つのSNPの遺伝子型を網羅的に調べる。GWASはGenome-Wide Association Studyの略。
次世代シークエンス技術または第三世代シークエンス技術を用いて、全ゲノムDNAを鋳型として配列を解読すること。この配列解読によって、全ゲノム長の数倍~数十倍の総塩基数に相当するショートリードまたはロングリードデータが生成される。構造多型の検出には、ショートリードでゲノム長の10~30倍、ロングリードで10倍以上の全ゲノムシークエンスデータを要する。
[2] 構造多型(SV)
ゲノムの個人間の違いのうち、50bp以上の大きさの変異。変異のパターンに応じて、欠失、挿入、重複、逆位、転座などに分類されるが、これらが混在した複雑なパターンを示す構造多型も存在する。通常、塩基対数の小さい構造多型ほど数が多いが、染色体レベルで起こる大きなサイズの構造多型も存在する。SVはStructural Variationの略。
[3] 遺伝子型
ある遺伝子座で個人が持つ遺伝子変異のこと。どちらかの親から一つの変異を受け継いでいる場合ではヘテロ遺伝子型となり、両親から同じ変異を受け継いでいる場合にはホモ遺伝子型となる。
[4] バイオバンク?ジャパン(BBJ)
日本人集団27万人を対象とした生体試料のバイオバンクで、東京大学医科学研究所内に設置されている。理化学研究所が取得した約20万人のゲノムデータを保有する。オーダーメイド医療の実現プログラムを通じて実施され、ゲノムDNAや血清サンプルを臨床情報とともに収集し、研究者へのデータ提供や分譲を行っている。
[5] 量的形質
身長や体重など連続的、量的に変化する形質。疾患の有無などのbinary形質と区別される。
[6] 欠失
構造多型のタイプの一つで、ゲノム配列の一部が失われた形態。挿入と並び、構造多型の中では最も多く存在する。
[7] 挿入
構造多型のタイプの一つで、ゲノム配列の特定の位置に別の配列が挿入された形態。挿入配列で最も多くあるものが、内在レトロ因子が挿入されたタイプで、ミトコンドリア配列やウイルスゲノム配列が挿入されたタイプもある。欠失と並び、構造多型の中では最も多く存在する。
[8] 重複
構造多型のタイプの一つで、ゲノム配列の一部の領域が重複して(2コピー以上)挿入された形態。欠失や挿入に比べて数は少ないが、重複された領域内に遺伝子が含まれる場合、通常と異なる遺伝子発現パターンを示すことが多いため、遺伝子機能の喪失を引き起こす欠失と同様、疾患との関わりが多く報告されている。
[9] 逆位
構造多型のタイプの一つで、ゲノム配列の一部が通常と逆方向に変換されている形態。構造多型のタイプの中で最も数が少ない。
[10] 一塩基多型(SNV)
ゲノムの個人間の違いのうち、塩基配列上の1カ所の違い(置換)が一塩基多型と定義される。SNVはSingle nucleotide variantの略。SNVのうち、ある集団内での頻度が1%以上あるものをSNP(Single Nucleotide Polymorphism)と呼ぶ。
[11] リード
DNAの配列決定(シークエンシング)によって得られるDNA断片の配列情報。次世代シークエンシング技術で得られるリードは、通常100~200bpのショートリード断片であり、ヒトゲノム解読の場合、数億~10億本のリードを得る。第三世代シークエンシング技術では、平均7~10Kb(7,000~10,000bp)のロングリードが得られる。
[12] リファレンス配列
ある生物種のゲノム配列として、標準ゲノム配列として公開されているもの。ヒトでは、hg19やGRCh37などの総塩基数約3Gb(30億b)のゲノムリファレンスが公開されている。リファレンスにリードデータをアライメントすることにより、標準リファレンス配列と異なるDNA多型が検出される。
[13] アライメント
シークエンスリードをリファレンス配列上の合致する位置に対応付けすること。通常、ショートリードはbwaなどのアライメントツールを用いてアライメントし、得られたアライメントファイルを用いて構造多型を検出する。
[14] アノテーション
ゲノム上の遺伝子領域などに注釈付けを行うこと。遺伝子がどのゲノム位置にコードされているか、SVなどのゲノム変異がどの遺伝子領域と重なっているかなどの注釈付けがある。
[15] 1000人ゲノムプロジェクト
ヒトゲノムの遺伝的多様性を明らかにすることを目標として開始された国際共同研究プロジェクト。現在では2,500人以上のゲノム解析へ移行している。
[16] 参照パネル、インピュテーション
ヒトの場合、DNAマイクロアレイを用いて得られたデータには数十万のSNP情報が含まれるが、実際には数百万以上のSNPが存在する。アレイに含まれないSNPやインデルを推定する手法がインピュテーションである。通常多くのサンプルの全ゲノムシークエンスデータから得られたSNP遺伝子型情報を参照パネルとして、そのSNP遺伝子型の並び情報を基に遺伝子型推定を行う。
[17] SNPアレイデータ
SNP(Single Nucleotide Polymorphism)は一塩基多型(SNV)のうち、ある集団内での頻度が1%以上あるものを指す。SNPアレイデータは、DNAマイクロアレイを用いて得られるSNP遺伝子型データのこと。全ゲノムシークエンスデータを用いて得られるSNP遺伝子型よりも少ない遺伝子型情報しか得られないが、全ゲノムシークエンスよりも安価に解析できる。
[18] ゲノムワイド関連解析(GWAS)
疾患などの特定の形質を持った集団と持たない集団との間でSNP遺伝子型の頻度差の有意性を統計的に評価する手法。ゲノム全域にわたって一つ一つのSNPの遺伝子型を網羅的に調べる。GWASはGenome-Wide Association Studyの略。
共同研究グループ
ゲノム解析応用研究チーム
チームリーダー 寺尾知可史(テラオ?チカシ)
(静岡県立総合病院 臨床研究部 免疫研究部長、欧洲杯押注平台_2024欧洲杯网站-官网app 薬学部 ゲノム病態解析分野 特任教授)
研究員(研究当時) 小杉俊一(コスギ?シュンイチ)
(現 客員研究員、静岡県立総合病院 リサーチサポートセンター 遺伝研究部 研究員)
上級技師 冨塚耕平(トミヅカ?コウヘイ)
人材派遣 原田勝利(ハラダ?カツトシ)
基盤技術開発研究チーム
チームリーダー 桃沢幸秀(モモザワ?ユキヒデ)
東京大学大学院 新領域創成科学研究科
教授 鎌谷洋一郎(カマタニ?ヨウイチロウ)
(理研 生命医科学研究センター ゲノム解析応用研究チーム 客員主管研究員)
特任研究員 森崎隆幸(モリサキ?タカユキ)
チームリーダー 寺尾知可史(テラオ?チカシ)
(静岡県立総合病院 臨床研究部 免疫研究部長、欧洲杯押注平台_2024欧洲杯网站-官网app 薬学部 ゲノム病態解析分野 特任教授)
研究員(研究当時) 小杉俊一(コスギ?シュンイチ)
(現 客員研究員、静岡県立総合病院 リサーチサポートセンター 遺伝研究部 研究員)
上級技師 冨塚耕平(トミヅカ?コウヘイ)
人材派遣 原田勝利(ハラダ?カツトシ)
基盤技術開発研究チーム
チームリーダー 桃沢幸秀(モモザワ?ユキヒデ)
東京大学大学院 新領域創成科学研究科
教授 鎌谷洋一郎(カマタニ?ヨウイチロウ)
(理研 生命医科学研究センター ゲノム解析応用研究チーム 客員主管研究員)
特任研究員 森崎隆幸(モリサキ?タカユキ)

